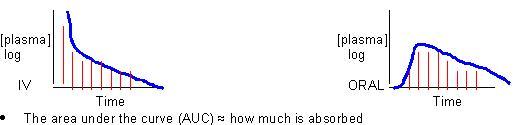Drugs get into the body by Absorption (get into the plasma)
The drug is distributed to the site of action
Unbound "free" drug is what has effects (bound drug exists in a equilibrium)
Tissue deposits can act as reservoirs
Drugs can be administered by ENTERAL: Oral (has the most variability), Sublingually, or Rectally
PARENTERAL: IV (intra-vascularly, can be intra-venous or intra-arterially), IM (intramuscularly), SC (subcutaneously), also by inhalation or intra-nasally.
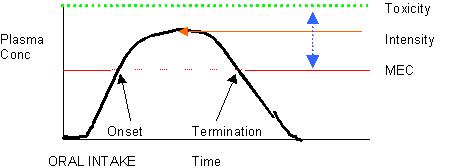
MEC – Minimal Effective Concentration
Intensity – is dependent on the peak plasma concentration
Toxicity – the level, once reached, when the drug is no longer therapeutic, it’s toxic!
Duration is dependent on the time between reaching the MEC and when the levels decay below the MEC.
Therapeutic range- is between the MEC and the Toxicity level
Absorption
Non-ionic passive diffusion, driven by the concentration gradient. Non-ionized
Þ lipid soluble
Active and Carrier mediated (facilitated diffusion) also can occur but rarely
Passive non-ionic diffusion depends on Weak Organic Acids (WOA’s) and Weak Organic Bases (WOB’s)
WOA: H+ + D-
« HD
When protonated the drug is neutral and absorbable
The rate of penetration = (permeability coefficient)(surface area)(concentration difference)
A pill’s composition determines where and when the pill disintegrates and dissolves into the GI liquid
Once through absorption the drug goes into the plasma where it is carried away. The blood flow removes the drug from the site of diffusion and keeps the concentration gradient going.
The surface area is important (the small intestine has a much greater surface area compared to that of the stomach)
Example Acetaminophen – pill disintegrates in the stomach (WOA), the rate of gastric emptying determines the time until peak plasma concentration is reached. To speed up absorption take Tylenol with ice water
With passive non-ionic diffusion
Þ more one takes, the more one absorbs
as concentration increases, rate of absorption increses in a linear fashion
With carrier (facilitated diffusion or active transport) there is a limit to the number of carrier/transporters, therefore the system can become saturated
as concentration increases, rate asymptotically approaches a maximum limit
Pharmacokinetics
The time it takes to get a drug to its site of action
With Oral administration: lag time, increasing concentration,
stays at peak (plateau), then decay.
MEC – Minimal Effective Concentration
Intensity – is dependent on the peak plasma concentration
Toxicity – the level, once reached, when the drug is no longer therapeutic, it’s toxic!
Duration is dependent on the time between reaching the MEC and when the levels decay below the MEC.
Therapeutic range- is between the MEC and the Toxicity level
Distribution
Drug moves around the body by Passive Non-ionic Diffusion
Distribution is a mathematical concept so one can relate something one can sample (e.g. [serum]) to tissue handling of a drug (which one can not sample); concentration = mass/volume
In plasma
Albumin binds WOA’s Drugs + Albumin becomes Dalb
The binding of Albumin keeps the bound form of the drug in the plasma (less drug gets into tissue)
a
1-acid glycoprotein – binds WOB’s, increased synthesis by the liver when sick. After a MI, concentration of a1-acid protein goes up.
Lipoproteins – can bind very lipid soluble drugs
Warfarin binds tightly to plasma albumin. Therefore Warfrin is confined to a small compartment. A "small volume of distribution" relative to the water space; Nortriptyline = WOB
In tissues
Lipid soluble drugs get into tissues and bind lipids and proteins within the tissues. (drug is sequestered in the tissue)
Acetaminophen and aminoglycosides do this. Tricyclic anti-depressants bind tightly to lipids in tissues.
Metabolism (elimination)
Phase 1
Lipid Þ "phase 1 step"Þ puts a H2O soluble part on the drug to render it more water soluble.
Cytochrome p450 is responsible for phase 1. (p450 is present in tissues but mainly in the liver)
The water soluble form can be activated or inactivated by this process
Phase 2
Makes the drug even more water soluble by adding a sugar to it (glucoronidated)
Reabsorption will occur by passive non-ionic diffusion (the pH of the urine will have an effect on how much drug is taken back up)
WOA’s and WOB’s are stripped off Albumin and
a 1-acid glycoprotein by a carrier at the end of the Proximal convoluted tubule and secreted into the lumen (urine); Penicillin is handled this way.
Since there are a limited amount of carriers, there can be competition between the drug and naturally occurring WOA’s that need to be excreted.
Pharmacokinetics
Most drugs behave such that as one increases concentration the velocity increases linearly "1st Order"; 1st order = proportional
Some drugs saturate (e.g. aspirin and phenytoin)
As one increases the dose, the plasma concentration rises linearly up to the saturation point. The plasma concentration is a mirror image of the saturation curve.
phenytoin (an anti-epileptic medication) has a therapeutic range of 10-20
mg/ml
since phenytoin’s KM
» 5 m g/mL if one doubles the dose into the therapeutic range the patient’s plasma level goes way up. Þ it saturates so most is in the "free" form
The half-life is how long it takes to get rid of half of the drug from the body.
Bioavailability
– How much drug is being absorbed?
The bioavailability of Oral = Fo; Fo = AUCoral / AUCIV assuming that AUCIV = 100%(ranges from 0-1)
Action
Knowing the action of the drug (exactly what it does) and at what concentration that effect is saturated, one can avoid giving excess medication and thereby limit the side effects.