Neoplasms of Bone and Cartilage
Osteochondroma
– exostosisChondromas
– benign tumors of hyaline cartilageChondrosarcoma
– group of tumors who all have the common feature of producing neoplastic cartilage|
ENCHONDROMA |
CHONDROSARCOMA |
|
Ceases to grow in adulthood |
Growing lesion |
|
Asymptomatic |
Painful |
|
Well defined border |
Poorly defined margin |
|
Regular ring like mineralization |
Irregular mineralization, poorly defined area |
|
No cortical destruction, thickening |
Cortical thickening or erosion |
Ewings Sarcoma
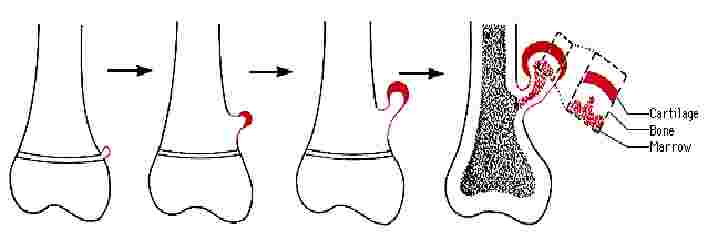
– a primary malignant small cell tumor of boneOsteosarcoma
– malignant mesenchymal (stromal) tumor in which the cancerous cells produce bone matrix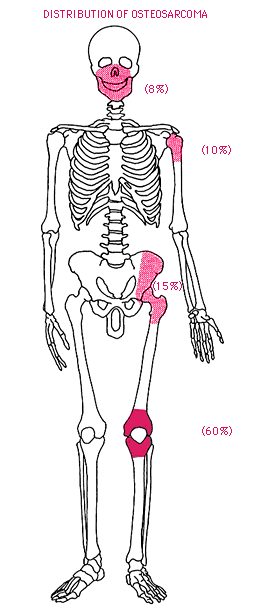
Metastatic Bone Disease
: Most common form of skeletal malignancy (osteosarcoma is most common primary cancer)