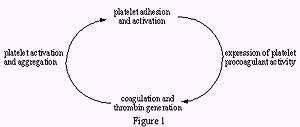
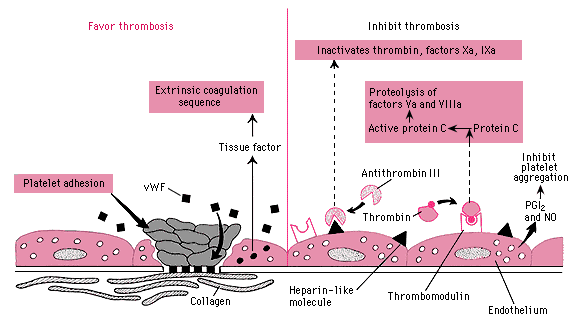
process by which platelets adhere to damaged endothelium or exposed connective tissue.
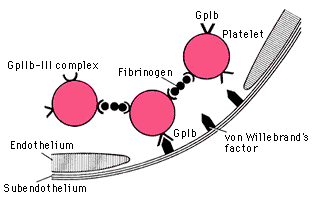
Adhesion: occurs through interactions between platelet adhesion receptors (GP 1b/IX/F and GP IIb/IIIa complexes), adherent platelets become activated
Þ secrete ADP Þ express fibrinogen receptors Þ activation and aggregation of additional platelets and formation of a platelet plug.
Activated platelets expose membrane phospholipids – provides a surface on which a series of reactions involving coagulation factors may occur (secondary hemostasis)
Thrombin: causes additional platelet activation and aggregation, cleaves fibrinogen to fibrin (serves as "mortar" which cements the platelet plug together
Clinical correlation:
Defect in primary hemostasis: primarily abnormal platelet function, tends to bleed immediately after trauma with difficulty in achieving initial hemostasis, bleeding often involves superficial sites (skin, mucous membranes).
vWF deficiency =
von Willebrand’s disease Ý bleeding times and (because vWF stabilizes factor VIII) Ý aPTT. Autosomal dominant.
GpIb deficiency =
Bernard-Soulier syndrome, autosomal recessive
GpIIb/IIIa deficiency
= Glanzmann’s thrombasthenia, auto recessive
Defect in secondary hemostasis: involves soluble coagulation factors, bleed at internal sites (hemarthrosis, retroperitoneal, intracranial bleeding)
Secondary Hemostasis: The Coagulation Cascade
– coagulation occurs on SURFACES, NOT in the fluid phase
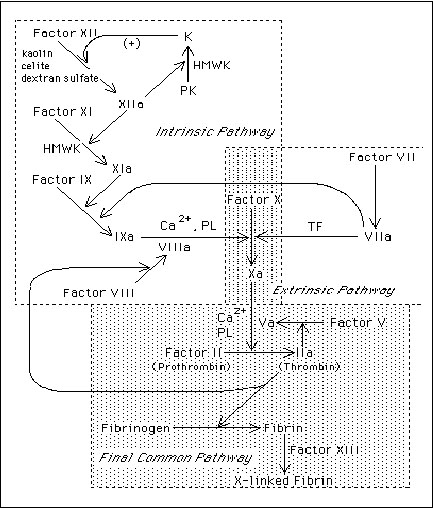
Intrinsic pathway
initiated by activation of factor XII (Hageman Factor) to activated Factor XII (XIIa)
Factor XII: capable of binding to negatively-charged surfaces. In vitro: glass, kaolin, celite, dextran. In vivo: components of basement membranes (collage, sulfatides, glycosaminoglycans)
Factor XIIa subsequently activates 2 proteins: Factor XI and prekallikrein (PK) to form XIa and kallikrein. Both proteins circulate in non-covalent bimolecular complexes with another protein High Molecular Weight Kininogen (HMWK)
HMWK: facilitates the activation of Factor XI and PK by binding negatively-charged surfaces in apposition to Factor XIIa
Kallikrein: accelerates activation of the intrinsic pathway by activating additional factor XII (a positive feedback loop)
Clinical Correlation:
Deficiency of Factor XII, prekallikrein, HMWK: do not bleed despite prolonged clotting times (in-vitro). Suggests that the intrinsic pathway in vivo does not have significant physiological importance.
Substrate for factor XIa is factor IX (Christmas factor)
Vitamin K dependent Coagulation factors:
Factors 2 (prothrombin),7,9,10
Vitamin K: required for the g -carboxylation of glutamic acid residues in the gla domains of the protein. The addition of a carboxyl group to glutamic residues provides them with a negatively charged surface allowing Ca2+ to bind which acts as a bridge to bind the protein to negatively charged phospholipid surfaces.
Clinical correlation:
Vitamin K deficiency is a common cause of prolonged prothrombin time. Most common in: ill patients with poor nutritional status taking systemic antibiotics.
Deficiencies seen in:
hemorrhagic disease of the newborn (Vitamin K does not cross placenta well)
Treatment: correction with vitamin K seen in 12 hrs
Factor IXa converts Factor X to Factor Xa. Reaction requires phospholipid, Ca2+ and Factor XIIIa
Tenase complex: Factor IXa, Factor XIIIa, phospholipid, Ca 2+
Phospholipid: serves as a negatively-charged surface on which factors assemble
Ca2+: serves as a bridge between factors IX and X, and the phospholipid
Factor VIII: serves as a cofactor for factor IX, activity greatly Ý by thrombin (IIa): + feedback loop
Clinical Correlation:
congenital deficiencies of factor VIII or IX results in Hemophilia A and B, respectively. Associated with bleeding severity proportional to the degree of deficiency. Circulating coagulation factor deficiency (i.e. deep muscle, joints, retroperitoneal, intracranial)
30% of normal coagulation factors required for normal hemostasis
Extrinsic Pathway
Involves both intra and extravascular elements (contrast to intrinsic where all components are found in the vasculature)
Tissue factor (tissue thromboplastin)
: lipoprotein consisting of a polypeptide membrane of many cells, produced by endothelial cells and vascular wall cells. Damage to tissues Þ Ý exposure of tissue factor
Factor VIIa: unclear what activates it : In vivo - factor VII, VIIa; In vitro – Factor IXa, Xa, XIIa
Complex of tissue factor, Factor VIIa has 2 actions: (1) converts factor X Þ Xa and (2) Factor IX Þ IXa (in presence of Ca2+)
The extrinsic pathway is also able to activate factor IX of the intrinsic pathway. This is currently believed to by the major route for coagulation pathway activation in vivo.
The Final Common Pathway
Newly formed Factor Xa forms prothrombinase complex
Prothrombinase complex:
complex of Factor Xa, phospholipid, Ca2+, Factor V and Factor II (prothrombin)
Action:
activation of prothrombin to thrombin; activation is 300,000x faster with complex than Factor Xa alone
Ca2+:
serves as a bridge between vitamin K factors (factors X, prothrombin) and the phospholipid surface
Factor V:
cofactor for Factor Va
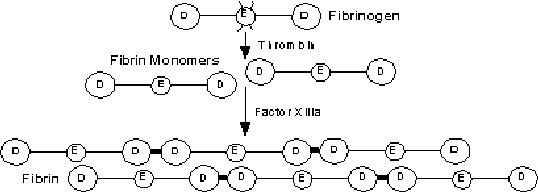
Thrombin
catalyzes the activation of fibrinogen to fibrin
Fibrinogen:
symmetric dimeric protein (a ,b ,g chain)
Thrombin:
cleaves small peptides (fibrinopeptides A and B) from a and b chains of fibrinogen Þ forms fibrin monomers
Fibrin monomers: associate laterally to form fibrin polymers which are linked by Factor XIII
Þ forms fibrin protofibrils Þ comprises fibrin clot and forms meshwork around platelets which aggregate around vascular defect. Mature fiber contains ~100 protofibrils
Regulation
– Coagulation cascade is regulated at several steps by naturally-occuring anticoagulant proteins
Antithrombin III
(AT III) – plasma protein which is a potent inhibitor of thrombin. Also Xa, IXa, XIa, and XIa. It’s interaction with thrombin and Factor Xa are of greatest importance. Action occurs on ALL cells wgere it is bound by heparan sulfate (a heparin-like glycosaminoglycan expressed on the surface of endothelial cells)
Deficiency:
Ý risk of thrombosis
Heparin:
Ý activity of AT III by 1000x. Drug binds to ATIII and alters its conformation (binds heparin sulfate on endothelial cells) – if ATIII deficient: multiple venous thrombosis before age 30
Clinical correlation:
Heparin mediates its anticoagulant effects through antithrombin III (therefore has limited efficacy AT III deficient patients)
Unfractionated heprin
: inactivates both thrombin and ATIII
Þ prlongs PTT time
Low molecular Weight heparin
: binds to AT III but NOT to thrombin Þ does NOT prolong PTT time
Thrombomodulin/ProteinC/ProteinS
Thrombomodulin: naturally-occurring anticoagulant protein expressed on the surface of vascular endothelial cells
Action:
binds thrombin, alters its specificity so that it no longer cleaves fibinogen by instead activates protein C to APC (activated protein C) that in turn inactivates Va and VIIIa (in the presence of S)
Protein C and Protein S:
Protein C in the presence of protein S (plasma cofactor) inactivates Factors Va and VIIa. They are anticoagulants
Clinical Correlation
: Protein C or S deficiency -
Ý risk for venous thrombosis.
Factor V leyden
: Mutation in cleavage site of Factor V renders it resistant to inactivation by Protein C leading to Ý risk for venous thrombosis; mutation in 3-5% of Caucasian population, and is most common cause of a congenital hypercoagulable state
coexistance of Factor V Leiden with protein C deficiency is further
Ý risk for venous thrombosis
Tissue factor pathway inhibitor
: forms a tight complex with Factor Xa
TFPI-Xa complex: inhibits the TF-Factor VIIa complex by feedback inhibition, anticoagulant effect
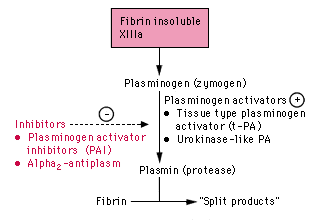
produced by vascular endothelial cells, most important activator in the vascular system (Clot buster)
Action:
binds tightly to fibrin and activates plasminogen to form plasmin.
Plasmin:
lyses the fibrin clot. Plasmin that escapes clot and cell-surface into the plasma are quickly inhibited by
a-2-macroglobulin and a-1-plasmin inhibitor.
u-PA:
extravascular and intravascular functions
Tissue factor pathway inhibitor (TFPI)
: discovered in 1989; forms a tight complex with Factor Xa that then inhibits the TF-VIIa complex by feedback inhibition – process occurs very rapidly after Xa generation and quickly squelches the activity of exposed tissue factor
deficiencies of TFPI in humans have not been demonstrated
in development for use as an anti-thrombotic
Lab measurement
2 primary lab tests: measure the function of intrinsic and extrinsic coagulation pathways in-vitro. Both depend on the final common pathway. Blood collected in tubes containing citrate. Citrate chelates Ca2+ so the sample won’t clot.
(1) Activated Partial Thromboplastin Time (aPTT).
Measure function of INTRINSIC coagulation pathway; indicates clotting factor deficiency (XII, PK, HMWK, XI, IX, VII, X, prothrombin, fibrinogen) or heparinization
Mechanism:
Plasma is exposed to kalin or celite to activate Factor XII. Phospholipid and Ca2+ are added to allow assembly of Factor IX/Ca/Factor VIII/Factor X complex as well as the prothrombinase complex. Mixture incubated at 37
° C and clotting time measured.
Clinical correlation:
PTT is prolonged by deficiencies of any of the intrinsic pathway clotting factors: XII, Prekallikrein, HMWK, XI, IX, VIII and factors X, prothrombin, fibrinogen
(2) Prothrombin time:
Measures function of EXTRINSIC coagulation pathway; indicates clotting factor deficiency (V, VII, X, Prothrombin, or fibrinogen) or DIC
Mechanism:
Ca2+, thromboplastin, added to patient palsma. Plasma incubated at 37
° C and clotting time measured.
Clinical correlation:
PT is prolonged by deficiencies of : Factor V, VII, and factors X, prothrombin or fibrinogen. Test most commonly prolonged in deficiency of Vitamin K. Preferentially prolongs in Disseminated intravascular coagulation (DIC)
Reported as INR = (patient PT/control PT) ISI (international standardized index): Due to differences in the thromboplastin reagent used by laboratories all reagents are standardized via the ISI so that all PT values are reported along with an INR
(3) Detection of coagulation factor Inhibitors:
coagulation assays may be prolonged by the presence of coagulation factor inhibitors. These inhibitors are antibodies directed against specific coagulation factors which neutralize the activity of that factor Þ causing a severe bleeding disorder.
Mixing study (Inhibitor screen)
: when a patient has a long PT or PTT, the next step is to note whether the deficiency is due to a deficiency in the clotting cascade or the presence of a coagulation inhibitor
Mechanism: patient’s plasma is mixed in a 1:1 ratio with the plasma of a normal donor. The PT or PTT is repeated. Since only 30-40% of normal activity is required to maintain PT or PTT within the normal range, if results are:
(1) Normal: than patient’s abnormal PT or PTT was due to a factor deficiency
(2) Prolonged: than the patient’s abnormal PT or PTT was due to a coagulation factor inhibitor because the inhibitor will continue to neutralze the specific coagulation factor in the donor plasma